丂峏偵丄僄僢僋僗慄夝愅傗妀帴婥嫟柭暘愅乮俶俵俼乯偵傛傞偨傫傁偔幙棫懱峔憿偺夝柧傕恑傫偱偍傝丄僐儞僺儏乕僞僔儈儏儗乕僔儑儞媄弍傪梡偄偨丄昗揑偨傫傁偔幙偺棫懱峔憿偵婎偯偔栻嵻愝寁乮俽俛俢俢丗丂俽倲倰倳們倲倳倰倕亅俛倎倱倕倓丂俢倰倳倗丂俢倕倱倝倗値乯傊偺婜懸傕崅傑偭偰偍傝傑偡丅偟偐偟丄廬棃偺僔儈儏儗乕僔儑儞媄弍偱偼丄寁嶼惛搙偑掅偄偐丄偁傞偄偼寁嶼帪娫偑偐偐傝偡偓傞偨傔丄幚尡傪戙懼偟偨傝丄偁傞偄偼幚尡傪曗彆偡傞廫暘側岠壥偑昁偢偟傕摼傜傟偰偄側偄偲偄偆栤戣偑偁傝傑偟偨丅
丂崱斒偺嫟摨尋媶偼丄偙偺傛偆側怴婯偑傫帯椕栻偺扵嶕傪栚揑偲偟丄俶俤俠偑撈帺偵奐敪偟偨僔儈儏儗乕僔儑儞媄弍偲擔杮壔栻偺峈偑傫嵻憂栻媄弍偵婎偯偒丄倝値丂倱倝倢倝們倧憂栻巟墖僔僗僥儉傪嫟摨偱峔抸偟丄幚嵺偺怴婯偑傫帯椕栻偺扵嶕傪嫟摨偱峴偭偨傕偺偱偁傝丄懡悢偺岓曗暔幙偺拞偐傜丄桳岠偲巚傢傟傞暔幙乮壔崌暔乯傪岠棪椙偔慖戰偡傞偙偲傪栚巜偟偨傕偺偱偁傝傑偡丅
丂擔杮壔栻偱偼丄崱屻丄偙偺偨傃敪尒偟偨怴婯壔崌暔偺峔憿嵟揔壔偲栻岠昡壙傪恑傔丄椪彴墳梡偵岦偗偰偦偺岠壥偲埨慡惈傪妋擣偟偰偄偔偲偲傕偵丄崱夞偺僔僗僥儉傪丄崱屻偺憂栻尋媶偺偨傔偺俬俿婎斦偲偟偰埵抲晅偗丄帺幮偺憂栻尋媶丒奐敪偺崅搙壔偵妶梡偡傞梊掕偱偁傝傑偡丅傑偨俶俤俠偱偼丄崱夞偺惉壥傪惢栻婇嬈岦偗偺倝値丂倱倝倢倝們倧憂栻巟墖僔僗僥儉偵偍偗傞拞妀媄弍偺堦偮偲偟偰妶梡偟丄棃擭搙偺惢昳壔偍傛傃帠嬈壔傪栚巜偟傑偡丅
嶲峫帒椏丂
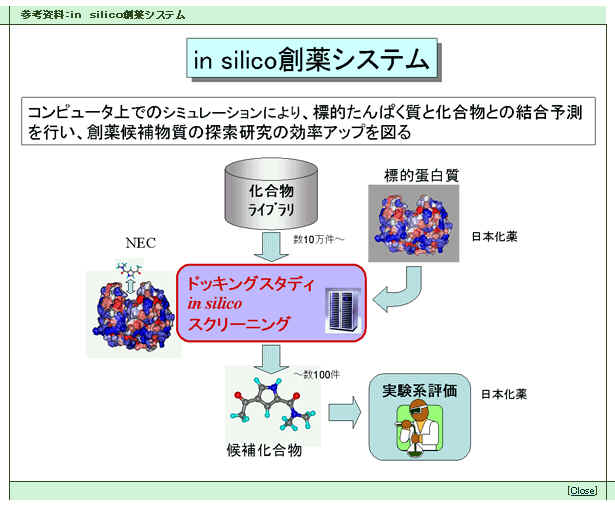
乮拲侾乯丂倝値丂倱倝倢倝們倧憂栻僔僗僥儉丗
丂僐儞僺儏乕僞傪梡偄偰丄偨傫傁偔幙側偳崅暘巕偺棫懱揑宍忬偵婎偯偄偰丄懡悢偺栻嵻岓曗壔崌暔偵偮偄偰丄偦偺偨傫傁偔幙偺偳偺晹埵偵偳偺傛偆偵寢崌偟偆傞偺偐乮僪僢僉儞僌儌乕僪乯丄傑偨偦偺寢崌偺嫮偝偑偳偺掱搙偱偁傞偺偐乮傾僼傿僯僥傿乯丄傪梊應偡傞偙偲偵傛偭偰丄廬棃偺倝値丂倴倝倴倧乮惗懱傪梡偄偨尋媶乯丄倝値丂倴倝倲倰倧乮帋尡娗撪偺尋媶乯偺曗彆偲偟偰丄岠棪揑側憂栻巟墖傪偡傞僔僗僥儉丅
乮拲俀乯丂億傾僜儞丒儃儖僣儅儞乮俹倧倝倱倱倧値亅俛倧倢倲倸倣倎値値乯曽掱幃丗
丂偨傫傁偔幙偲壔崌暔偺暋崌懱偺寢崌偺嫮偝偵懳偟偰丄廃埻偺悈傗僀僆儞偑偍傛傏偡惷揹椡偺岠壥傪悇嶼偡傞曽朄丅廃埻偺悈暘巕傪楢懕桿揹懱偵娙棯壔偟丄偨傫傁偔幙偲壔崌暔偺暋崌懱偵摥偔惷揹椡傪儊僢僔儏忬偺懡悢偺奿巕忋偱乮嵎暘乯嬤帡偟丄崅懍偵寁嶼丅悈暘巕傪桿揹懱偵嬤帡偟側偄応崌偵斾傋丄惛搙偼庒姳媇惖偵側傞偑丄崅懍壔偑壜擻偱偁傝丄戝検壔崌暔偐傜偺岓曗慖敳偵揔偟偰偄傞丅
乮拲俁乯丂暘巕摦椡妛朄丗
丂偨傫傁偔幙側偳偺暘巕傪峔惉偡傞尨巕偺尨巕妀偵摥偔憡屳嶌梡椡傪丄僶僱傗僋乕儘儞椡偺傛偆側娙扨側億僥儞僔儍儖娭悢乮暘巕椡妛儌僨儖乯偱嬤帡偟丄暘巕偺擬塣摦傪丄僯儏乕僩儞偺塣摦曽掱幃傪夝偄偰僔儈儏儗乕僔儑儞偡傞曽朄丅崱夞偼丄僄僱儖僊乕嵟彫壔寁嶼偵巊梡偟偨偑丄偙偺曽朄傪梡偄偰傛傝惓妋偵暘巕偺寢崌僄僱儖僊乕傪梊應偡傞偨傔偵偼丄侾愮枩僗僥僢僾乮侾侽僫僲昩乯偺寁嶼偑昁梫偲峫偊傜傟偰偍傝丄崅懍僴乕僪僂僃傾偵懳偡傞婜懸偑崅偄丅